
НевроМед
спеціалізований медичний центр
Епілептична енцефалопатія з тривалою спайк-хвильовою активністю уві сні
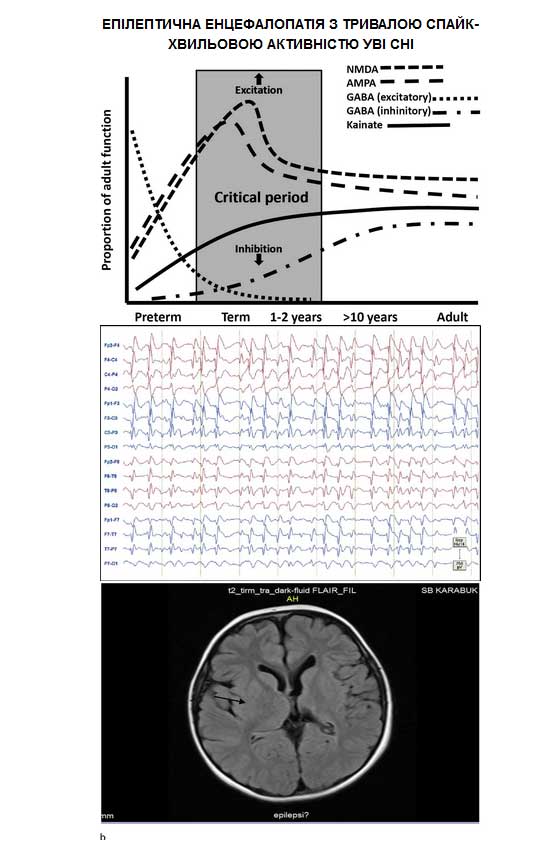
Епілептична енцефалопатія з продовженою спайк-хвильовою активністю уві сні (епілепсія з електричним епілептичним статусом в фазі повільного сну, або енцефалопатія з електричним епілептичним статусом в фазі повільного сну) є частково зворотною залежною від віку епілептичною енцефалопатією, для якої характерна тріада симптомів: продовжена спайк-хвильова активність уві сні (електричний епілептичний статус в фазу повільного сну), судоми і нейропсихологічні порушення.
Термінологія і питання класифікації
Більшість дослідників вважають, що поняття “електричний епілептичний статус в фазу повільного сну” (ESES — від англ. electrical status epilepticus in sleep) і “продовжена спайк-хвильова активність уві сні” (CSWS — від англ. continuous spikes and waves during sleep) є синонімами. Деякі автори вважають, що поняття “електричний епілептичний статус в фазі повільного сну” відображає тільки зміни на ЕЕГ, а “продовжена спайк-хвильова активність уві сні” являє собою не тільки аномалії ЕЕГ, а й нейропсихологічні порушення. У літературі, присвяченої даному питанню, фігурують обидві точки зору.
У 2009 році Комісія по класифікації та термінології в області епілепсії запропонувала включити в епілептичну енцефалопатію з продовженою спайк-хвильовою активністю уві сні (CSWS) синдром Ландау-Клеффнера. Цю точку зору підтримав і сам першовідкривач електричного епілептичного статусу уві сні С.А. Tassinari. Він вважає, що синдром Ландау-Клеффнера є клінічним варіантом ESES, а різні варіанти нейропсихологічних порушень обумовлені різною локалізацією епілептичного фокусу (при синдромі Ландау-Клеффнера — у скроневій частці, при ESES — переважно лобною локалізацією). Видається, що це не зовсім точно. Добре відомо, що існують пацієнти з синдромом Ландау-Клеффнера, що не мають судом або явищ CSWS. Крім того, найяскравішим клінічним проявом синдром Ландау-Клеффнера є те, що дитина перестає розуміти звернену до неї мову (вербальна агнозія), і тільки потім, в міру перебігу хвороби, може порушуватися експресивна мова. У дітей з енцефалопатією з CSWS, як правило, в першу чергу страждає саме експресивна мова при відносно збереженому її розуміння. Необхідні подальші спостереження і дослідження для того, щоб зрозуміти наскільки епілептична енцефалопатія з CSWS і синдром Ландау Клеффнера є самостійними епілептичними синдромами, або все-таки вони є частинами одного і того ж континууму. Очевидно, те ж саме стосується і такого рідкісного епілептичного синдрому, як атипова доброякісна епілепсія дитинства (або синдром псевдо-Леннокса), клінічні і ЕЕГ-характеристики якого схожі з епілептичною енцефалопатією з продовженою спайк-хвильовою активністю уві сні.
Поширеність
Точних даних про поширеність епілептичної енцефалопатії з CSWS немає. Вважається, що частота синдрому в епілептологічних клініках становить 0,5% всіх випадків епілепсії у дітей. Пацієнти з епілептичною енцефалопатією з CSWS становлять 1–2% осіб, що зазнають нейрохірургічних втручань з приводу резистентної епілепсії. Можливо, що переважають хлопчики (62%). Даний синдром виникає у дітей, хоча є окремі вкрай рідкісні його випадки у дорослих.
Етіологія
Досі вважається недостатньо вивченою. Можна виділити дві основні групи причин — ранні за термінами розвитку пошкодження головного мозку і генетичні причини. Приблизно в 1/3 пацієнтів виявляються зміни на МРТ головного мозку — локальна або дифузна атрофія кори, поренцефалія, різні кортикальні мальформації. Підкреслюється, що значна частина пацієнтів має перинатальні ураження судинного походження. В окремих роботах підкреслюється значення перинатального ураження таламуса.
Генетичні чинники мало вивчені. Сімейна обтяженість по судомах, включаючи фебрильні, відзначається у 10–15% пацієнтів. У окремих хворих з даною епілептичною енцефалопатією описані варіації повторів копій генів і мутацій, що мають різну локалізацію (дуплікація Xp11.22-p11.23, мікроделеція 8q12.2q13 та ін.). Добре відома і детально описана еволюція епілептичної енцефалопатії з CSWS з доброякісних фокальних епілепсій дитинства (здебільшого з доброякісною фокальною епілепсією з центро-темпоральними спайками, рідко — з доброякісною потиличною епілепсією з раннім початком). До факторів ризику розвитку CSWS при роландовій епілепсії відносяться ранній початок хвороби (до 4 років), відсутність позитивного ефекту від призначення протиепілептичних препаратів і наявність мультифокальних епілептиформних розрядів на ЕЕГ. Саме тому при появі нових типів нападів, поведінкових, когнітивних або мовних порушень, а також частих синхронізованих епілептиформних розрядів дітям з доброякісною фокальною епілепсією дитинства показано проведення ЕЕГ уві сні. J. Lemke і співавт. в 2013 р. виявили мутацію в гені GRIN2A в 4,9% випадків пацієнтів з роландовою епілепсію і в 17,6% випадків пацієнтів з CSWS. Ген GRIN2A кодує NR2A-cубодиницю NMDA-рецепторів. Етіологічну роль генетичних факторів у розвитку CSWS ще варто уточнити.
У дослідженнях U. Kramer і співавт. 37% всіх випадків епілептичної енцефалопатії з CSWS складала атипова еволюція доброякісних епілепсій, 17% — епілепсія при дитячих церебральних паралічах; 17% — при гідроцефалії, в 3% — внаслідок вад розвитку головного мозку. Приблизно в 1/3 всіх випадків не вдається виявити структурних дефектів на МРТ головного мозку і обтяженого анамнезу щодо доброякісних фокальних епілепсій. У деяких дослідженнях співвідношення випадків з невідомою етіологією і симптоматичних випадків становило відповідно 35 і 65%.
Патогенез синдрому
Механізми розвитку синдрому також вивчені недостатньо. Існує гіпотеза про те, що його виникнення пов’язане з гіперактивністю кортико-таламо-кортикальної системи і з недостатньою функцією ретикулярної формації, яка стримує цю активність. Синдром розвивається в період активного синаптогенезу, аксонодендритного розгалуження і формування функціональних систем головного мозку. Припускають, що епілептична активність (CSWS) порушує цей процес, можливо, провокуючи розвиток неправильно функціонуючих синапсів.
В області патогенезу синдрому залишається неясним питання — чому у одних пацієнтів (наприклад, з однаковими структурними дефектами мозку) розвивається CSWS, а в інших — ні.
Безпосередній механізм, що генерує CSWS — вторинна білатеральна синхронізація. Як відомо, остання є енцефалографічним паттерном, що складається з послідовності фокальних спайків, полі-спайків або спайк-хвильових комплексів (рідше — повільних хвиль), за якими випливає спалах білатеральної, синхронної і симетричної спайк-хвильової активності, поширеної на обидві півкулі. Вторинна білатеральна синхронізація є результатом швидкого поширення фокального приступного розряду через мозолисте тіло і спайку гіпокампу. Цікаво, що такі функціональні методи, як МРТ головного мозку і позитронно-емісійна томографія (ПЕТ), у дітей з CSWS під час нападу демонструють збільшення метаболізму і/або кровоплину в епілептичному фокусі з паралельним їх зниженням в прилеглих і далеко розташованих відділах головного мозку, тобто патологічний процес носить досить поширений характер.
Клінічні прояви
Початок епілептичних нападів зазвичай припадає на вік від 2 місяців до 12 років (пік дебюту епілепсії — від 4 до 5 років). Вважається, що у пацієнтів зі структурними змінами головного мозку епілепсія починається раніше — у віці близько 2 років. CSWS розвивається через 1–2 роки після початку нападів (від 3 до 14 років).
У перебігу синдрому виділяють чотири стадії:
1. “Прихована” стадія — від народження до початку епілептичних нападів. Психомоторний і розмовний розвиток дитини може бути нормальним або характеризуватися легкою затримкою.
2. Продромальна стадія — від початку епілептичних приступів до виявлення CSWS. Характерні нечасті фокальні (прості або складні) переважно нічні епілептичні напади, іноді вторинно-генералізовані, в 40% випадків — нічні геміклонії. Як правило, у дитини є тільки один, рідко — два типи нападів. Психо-мовний розвиток дитини в цей період не погіршується. Але вже на цій стадії на ЕЕГ реєструються мультифокальні епілептиформні розряди, можливі також бісинхронні генералізовані спайк-хвильові розряди.
3. Гостра стадія — від розвитку психо-мовного регресу до припинення нападів. Вона настає через 1–2 роки від моменту першого епілептичного нападу. Відзначаються збільшення числа нападів, порушення психічного розвитку; в цей же час виявляється CSWS на ЕЕГ. У одного пацієнта можуть спостерігатися як однотипні напади, так і найрізноманітні, включаючи геміфаціальний, геміконвульсивні, атонічні, вторинно-генералізовані тоніко-клонічні, атипові абсанси, негативний міоклонус. Тонічні напади (якщо їх наявність показана на ЕЕГ) не характерні і є критерієм виключення цього синдрому. Їх наявність змушує задуматися про синдром Леннокса-Гасто. Можливий розвиток безсудомного епілептичного статусу. У 90% пацієнтів відзначається високе число приступів (багато протягом одного дня). Раптово розвиваються і швидко прогресують нейропсихологічні порушення. Їх характер визначається переважною локалізацією епілептоформної активності. Лобна локалізація викликає когнітивні рушення і порушення конструктивної діяльності до розвитку мовних порушень. Формується так звана лобна психіка — розгальмованість, гіперактивність, агресивність, дратівливість, риси аутизму, ажитація у поєднанні з розладами пам'яті та уваги. Епілептоформна активність здебільшого скроневої локалізації викликає перш за все мовні порушення, переважно — експресивну афазію (на відміну від вербальної агнозії при синдромі Ландау-Клеффнера). В період активного перебігу синдрому у пацієнтів виявляються рухові розлади: атаксія, геміпарез, диспраксія. У окремих пацієнтів спостерігають епілептоформний оперкулярний синдром (слиновиділення, дизартрія, слабкість мімічної мускулатури і язика).
4. Резидуальна стадія (після зникнення епілептичних нападів) — починається в термін від місяців до 2–7 років з моменту початку нападів і по суті є спонтанною клініко-лабораторною ремісією синдрому. ЕЕГ поступово нормалізується, нейропсихологічний статус також покращується, але психо-мовний розвиток не досягає середньої вікової норми. Багато дітей страждають тяжкими мовними порушеннями і розумовою відсталістю.
Типова еволюція синдрому за вказаними стадіями у деяких пацієнтів може бути відсутньою. Іноді у хворих немає нападів, але є поведінкові порушення, порушення мови і CSWS. Дослідження показують, що у значної кількості пацієнтів (17%) CSWS була до початку епілептичних нападів, а напади з'являлися через 2–4 роки, правда, при цьому діагноз CSWS ставився, якщо епілептиформна активність займала не менше 30% періоду запису сну. Крім того, існують окремі пацієнти, у яких є CSWS і епілептичні напади, але немає поведінкових, мовних і інтелектуальних порушень. У такій ситуації не слід беззастережно вірити батькам пацієнта, а провести дитині нейропсихологічне тестування, яке може виявити неочевидні для родичів порушення.
ЕЕГ-діагностика синдрому
Міжприступна ЕЕГ до розвитку CSWS характеризується фокальними або мультифокальними (у 2/3 пацієнтів) розрядами, частіше — лобно-скроневої або центрально-скроневої, рідше — тім’яно-потиличної локалізації. За морфологією вони нагадують доброякісні епілептиформні розряди дитинства. У 80% випадків відзначаються короткі дифузні генералізовані комплекси гостра-повільна хвиля з частотою 1–3 Гц, дуже часто — з чіткими фокальними спайками на початку розряду. Під час другої фази синдрому ЕЕГ в стані бадьорості має той же характер, але зміни більше виражені (епілептиформні розряди зустрічаються частіше). На ЕЕГ у сні розвивається продовжена або майже продовжена епілептиформна активність у вигляді білатеральних синхронних комплексів гостра-повільна хвиля з частотою 1,5–2 Гц (іноді 3–4 Гц). Амплітуда розрядів переважає в передніх відділах мозку. C. Tassinari і співавт. так описували феномен ESES: “Як тільки пацієнт засинає, появляються тривалі білатеральні дифузні повільні спайки і хвилі, які проходять через всі стадії повільного сну”. Міжнародна протиепілептична ліга пропонує замінити термін “Фаза повільного сну” на термін “фаза NREM-сну” (від англ. non-REM — без швидких рухів очей). У пропозиціях за новою класифікацією епілептичних синдромів йдеться про те, що CSWS з'являється “як тільки пацієнт засинає і триває протягом усіх стадій NREM-сну, переривається в REM-сні, потім відновлюється знову”. В REM-фазі (від англ. — з швидкими рухами очей) зміни аналогічні ЕЕГ, записаній в стані бадьорості. Кількісно спайк-хвильова активність описується так званим спайк-хвильовим індексом, який розраховується як загальна сума всіх спайк-хвиль (хв.) помножена на 100 і поділена потім на тривалість повільного сну (хв.). Згідно класичних уявлень, для того, щоб констатувати наявність ESES/CSWS, спайк-хвильовий індекс повинен становити не менше 85% NREM-сну. Вважається, що значення індексу вище в першому циклі сну (95–100%), ніж в наступних (70–80%).
Проте існують дослідження, які показують, що більш низькі значення спайк-хвильового індексу (менше 80%) асоційовані з нервово-психічним регресом. Тому, на жаль, існує значне різноголосся щодо того, який саме спайк-хвильовий індекс відповідає продовженій спайк-хвильовій активності уві сні (CSWS або ESES). За даними різних авторів, спайк-хвильовий індекс при цьому синдромі коливається від 25 до 90%, а деякі автори взагалі обмежуються лише фактом “значної активації розрядів уві сні”.
Ситуація ускладнюється тим, що методи підрахунку спайк-хвильового індексу також варіюють: можна рахувати індекс в NREM-фазу протягом всього нічного сну, або в кожному циклі NREM-сну, або в перші 30 хв. NREM-сну в перший і останній цикли сну, або в NREM-фазу в першому циклі сну і ін. Крім того, існує досить широка варіативність локалізації CSWS. Початково ESES описувався, як дифузний паттерн. Але в останні роки все частіше стали писати про те, що розряди уві сні можуть бути асиметричними, унілатеральними або переважно фокальними, і це не є критерієм виключення СSWS. Так, C. Tassinari описує як типову для ESES ЕЕГ з “дифузним або більш менш унілатеральним електричним епілептичним статусом”.
Такі складності у визначенні продовженої спайк-хвильової активності викликали необхідність досягнення консенсусу за критеріями її діагностики. У 2009 р. в журналі “Epilepsia” було надруковано “Керівництво по ЕЕГ при енцефалопатії, асоційованій з ESES / CSWS у дітей”. В ньому йдеться про те, що значна активація розрядів під час NREM-сну (іноді в REM-сні) зі спайк-хвильовим індексом, що становить не менше 50%, говорить про можливість існування CSWS. Підкреслюється, що епілептиформна активність на ЕЕГ під час бадьорості і сну може бути фокальною, мультифокальною, унілатеральною, асиметричною або симетричною білатеральною, а також дифузною. Паттерн CSWS може бути пролонгованим, фрагментованим або періодичним. У інструкції також відзначається, що не тільки епілептиформна активність під час бадьорості і сну, але й повільно-хвильова активність і порушення архітектоніки сну грають важливу роль в розвитку клінічних симптомів. Нормальні патерни ЕЕГ у сні (сонні веретена, К-комплекси, вертексні спайки) відмінні тільки в NREM-фазі, якщо CSWS фрагментована. Для наукового вивчення CSWS показаний 24-годинний відео-ЕЕГ-моніторинг (його можна проводити амбулаторно). Можна проводити полісомнографію. Спайк-хвильовий індекс краще оцінювати в перший і останній цикли сну. Деякі дослідники для пошуку клініко-енцефалографічних кореляцій розробляють спеціальні шкали. Так, Sheltens-de Boer пропонує наступну бальну оцінку наявності і ступеня вираженості CSWS: 0 балів — відсутність розрядів; 1 бал — 0–20%, 2 бали — 20–50%, 3 бали — 50–85%, 4 бали — понад 85% NREM-сну зайнято спайк-хвильовою активністю. У інструкції згадується про те, що для клінічних цілей запис відео-ЕЕГ (або просто ЕЕГ при неможливості проведення відео-ЕЕГ-моніторингу) може бути менше 24 год — в такому випадку пишеться ЕЕГ в стані бадьорості і короткого денного сну після депривації останнього. Є також рекомендація по частоті проведення ЕЕГ на фоні лікування: з науково-дослідницькою метою — перший раз через 2 тижні, потім через 4 тижні; з клінічної метою — якщо є підозра на рецидив або сумніви в клінічних зрушеннях.
Для синдрому характерна спонтанна еволюція паттерну CSWS. Навіть якщо пацієнт не отримує терапії або терапія неефективна, все одно протягом наступних років відбувається зменшення активності — розряди стають коротшими і рідшими, більш фрагментованими, більш помітні нормальні патерни сну. Можуть довго зберігатися фокальні комплекси гостра-повільна хвиля. Нормалізація ЕЕГ відбувається через роки — в середньому у віці близько 9–11 років, іноді після 15 років.
Лікування енцефалопатії з CSWS
Основною метою лікування синдрому є ліквідація або значне зменшення епілептиформної активності на ЕЕГ. Лікування нападів є також важливим, але не їх наявність визначає несприятливий прогноз синдрому. На жаль, ті препарати, які здатні переривати епілептичні напади, часто малоефективні щодо CSWS, а ті, які які в даному випадку більш ефективні, як правило, використовуються емпірично (точні їх дози і тривалість терапії не визначено). Подвійні сліпі дослідження по ефективності протиепілептичних препаратів (ПЕП) і гормонів відсутні, що пов’язано з відносною рідкістю синдрому і з його поліетіологічністю. Більшість робіт по ефективності терапії ґрунтуються на невеликій кількості спостережень і носять ретроспективний характер. Проте можна виділити основні принципи терапії.
Не застосовуються ПЕП, які можуть посилити вторинну білатеральну синхронізацію на ЕЕГ і тим самим викликати агравацію перебігу епілепсії. До них відносяться карбамазепін, окскарбазепін і фенітоїн.
Як правило, базисним препаратом, особливо на ранніх стадіях синдрому (до розвитку CSWS), є вальпроат. При відсутності позитивної динаміки (не тільки по відношенню до нападів, але й щодо поширеності розрядів на ЕЕГ) оптимальною є комбінація вальпроату і етосуксиміду. Якщо препарати добре сприймаються, вони можуть застосовуватися в досить високих дозах — до 50–60 мг/кг/добу, що виходить за рамки зазначеної в анотації максимальної добової дози етосуксиміду .За даними деяких авторів застосування даної комбінації в добових дозах ПЕП, що не перевищували допустимі, у 29% пацієнтів призводило до клініко-енцефалографічної ремісії.
Ефективним щодо CSWS у деяких пацієнтів може виявитися такий препарат, як сультіам, але в більш високих, ніж зазвичай, добових дозах (до 20 мг/кг).
З відносно нових ПЕП ефективним може виявитися леветирацетам, який застосовується зазвичай як додатковий (другий) препарат, в добових дозах 30–60 мг/кг. Він може вплинути як на епілептичні напади, так і на електричний епілептичний статус сну навіть у тих пацієнтів, які виявилися резистентні до бензодіазепінів.
Перераховані вище ПЕП і їх комбінації не забезпечують високу ефективність в лікуванні CSWS. Тому довго затримуватися на кожному етапі терапії не є можливим. Більш ефективними методами медикаментозного лікування є застосування бензодіазепінів і гормонів. Іноді, коли лікарі стикаються з різким регресом розвитку у дитини (наприклад, з різким погіршенням мови, пам'яті та уваги), тяжким перебігом нападів, необхідно відразу вдаватися до гормональної терапії або бензодіазепіну. Досить часто є пацієнти, які безуспішно спробували самі різні ПЕП, і тоді бензодіазепіни або гормональна терапія призначаються в якості додаткового третього медикаменту.
У міжнародній практиці для переривання CSWS досить широко використовуються бензодіазепінові препарати, зокрема клобазам перорально в середній добовій дозі 0,5–1 мг/кг/добу або ректальні форми бензодіазепінів. Останні стараються призначати коротким курсом протягом 3–4 тижнів і їх часто поєднують з вальпроатом. Як правило, терапія бензодіазепінами добре переноситься. На жаль, далеко не завжди терапія клобазамом може проводитися таким коротким курсом, багато пацієнтів при спробі зниження дози і відміни препарату дають рецидив електричного статусу у сні. У деяких пацієнтів з CSWS використовується клоназепам, але його застосування, на відміну від клобазаму, супроводжується вираженою сонливістю і м'язовою гіпотонією, тому він виявляється ефективним і добре переносимим тільки у окремих пацієнтів.
Альтернативою застосуванню бензодіазепінів є гормональна терапія, але її початкові етапи через можливі побічні ефекти повинні проводитися в стаціонарі. Оптимальним варіантом є перебування пацієнта в денному стаціонарі. Гормональна терапія епілепсії з епілептичним статусом в фазу повільного сну дозволяє домогтися ремісії швидше, ніж при застосуванні звичайної протиепілептичної терапії, що може сприяти більш швидкому відновленню когнітивних функцій пацієнтів і скоротити число візитів при подальшому амбулаторному спостереженні. Було показано, що преднізолон (добова доза 2–5 мг/кг), метилпреднізолон (20 мг/кг протягом 3 днів) і адренокортикотропний гормон (в добовій дозі 80 індивідуальних од.) можуть перервати CSWS і поліпшити пізнавальні функції. При досягненні ефекту (оцінюється по позитивній динаміці на ЕЕГ, динаміці мови і поведінки, а також по зменшенню частоти і ступеня тяжкості нападів) рекомендується поступове зменшення добової дози гормонів. Оптимальна тривалість гормональної терапії не визначена, але в цілому вважається, що вона не повинна бути менше 6 місяців. В останні роки стали описуватися досить довготривалі курси гормонів — до 2 років, але при такій тривалій терапії після ліквідації CSWS застосовуються підтримуючі невисокі дози. I. Fernandez і співавт. в якості гормональної терапії призначали гідрокортизон за наступною схемою: 5 мг/кг/добу 1-й місяць, 4 мг/кг/добу — 2-й місяць, 3 мг/кг/добу — 3-й місяць і 2 мг/кг/добу — наступні 9 місяців, потім повільне (протягом 9 місяців) зменшення дози і відміна через 21 місяць лікування. При рецидивах CSWS рекомендується повторення гормональної терапії, при цьому допускається, що вона може розтягнутися на роки.
У окремих пацієнтів з епілептичною енцефалопатією з CSWS може виявитися ефективною кетогенна дієта і внутрішньовенне введення високих доз імуноглобулінів.
Не слід забувати і про таку можливість, як нейрохірургічне втручання, особливо у тих пацієнтів, в яких CSWS обумовлена фокальними дисплазіями кори.
Прогноз
Епілептичні напади припиняються в віці від 10 до 15 років. Припинення нападів може бути одночасно зі зникненням паттерна CSWS на ЕЕГ, передувати цьому зникненню або слідувати за ним. Загальна тривалість активного перебігу нападів варіює від 4 до 17 років. Спонтанна еволюція нейропсихологічних порушень відбувається після 10 років. Ступінь вираженості нервово-психічних порушень залежить від віку розвитку CSWS і тривалості її існування. Чим раніше розвивається CSWS і чим довше вона існує, тим гірший прогноз. Більшість пацієнтів не повертаються до нормального рівня уваги, когнітивних і мовних функцій. Тільки одна чверть мають прийнятний рівень інтелекту, уваги і мови. Це ті пацієнти, у яких був нормальний інтелект і неврологічний статус до початку епілепсії і коротка тривалість CSWS.
Література
1. Hughes JR. A review of the relationships between Landau-Kleffner syndrome, electrical status epilepticus during sleep, and continuous spike-waves during sleep. Epilepsy Behav. 2011 Feb;20(2):247-53.
2. Loddenkemper T, Fernández IS, Peters JM. Continuous spike and waves during sleep and electrical status epilepticus in sleep. J Clin Neurophysiol. 2011 Apr;28(2):154-64.
3. Nickels K, Wirrell E. Electrical status epilepticus in sleep. Semin Pediatr Neurol. 2008 Jun;15(2):50-60.
4. RamachandranNair R. Encephalopathy Associated with Electrical Status Epilepticus of Sleep (ESES): A Practical Approach. Indian J Pediatr. 2020 Dec;87(12):1057-1061.
5. Samanta D, Al Khalili Y. Electrical Status Epilepticus In Sleep. 2021 Jul 7. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021 Jan.
6. Sánchez Fernández I, Loddenkemper T, Peters JM, Kothare SV. Electrical status epilepticus in sleep: clinical presentation and pathophysiology. Pediatr Neurol. 2012 Dec;47(6):390-410.
7. van den Munckhof B, van Dee V, Sagi L, Caraballo RH, Veggiotti P, Liukkonen E, Loddenkemper T, Sánchez Fernández I, Buzatu M, Bulteau C, Braun KP, Jansen FE. Treatment of electrical status epilepticus in sleep: A pooled analysis of 575 cases. Epilepsia. 2015 Nov;56(11):1738-46.
8. Veggiotti P, Pera MC, Olivotto S, De Giorgis V. How to Manage Electrical Status Epilepticus in Sleep. J Clin Neurophysiol. 2016 Feb;33(1):3-9.
9. Veggiotti P, Pera MC, Teutonico F, Brazzo D, Balottin U, Tassinari CA. Therapy of encephalopathy with status epilepticus during sleep (ESES/CSWS syndrome): an update. Epileptic Disord. 2012 Mar;14(1):1-11.